
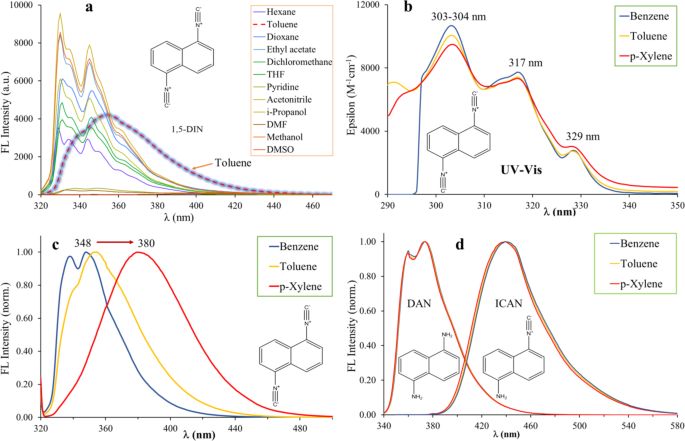
Proof of aromatic complex formation with 1,5-diisocyanonaphthalene (1,5-DIN)
Previously we demonstrated that 1,5-diisocyanonaphthalene (1,5-DIN) has the potential to become a versatile and powerful fluorescent probe30. Due to its symmetric structure, including two strongly electron withdrawing isocyano groups on the naphthalene core at 1,5 positions, the shape and the position of the emission peaks change little in solvents of different polarity (Fig. 1a). In most solvents, the original double peak with maxima at approximately 330 and 345 nm can be identified. However, in toluene the spectrum changes dramatically. Instead of two peaks, only one broad peak appears at 354 nm with a shoulder at 340 nm. The broadening and significant redshift of the emission peak may suggest the presence of specific interactions such supposed as π-π stacking. To test this assumption, UV-Vis and steady-state emission spectra were recorded in common aromatic solvents, such as benzene, toluene and p-xylene, as shown in Fig. 1b-c. The UV-Vis spectra (Fig. 1b) are almost identical in benzene, toluene and p-xylene. The most intense absorption peak is found at 303–304 nm with molar extinction coefficients (ε) varying between 9500 and 10,700 M-1cm-1. Two lower intensity peaks are also found at lower energies, at 317 and 329 nm, respectively. For fluorescence studies we used the absorption band at 317 nm as excitation wavelength to reduce the possible overlap with the absorption of the pure aromatic solvents near 300 nm. Unlike the absorption spectra, the emission spectra differ significantly. In benzene, the original double peak structure is retained, however, the two peaks have almost the same intensity, broadening of the peaks and a slight redshift of 8 and 3 nm (from 330 to 338 nm and from 345 to 348 nm) can also be observed. More pronounced bathochromic shift is identified in the case of toluene. Only one broad peak at 354 nm (∆λem, max=9 nm) with a left shoulder at 341 nm appears, it may be the remnant of the higher energy peak of the doble peak structure. Upon the addition of p-xylene, the emission spectrum changes significantly, as only single broad charge transfer (CT) like emission band is found with a maximum of 380 nm, which is red-shifted by 35 nm and 26 nm relative to those observed in MeCN and in toluene, respectively. Similarly, to the bathochromic shift of the emission maximum, the full width at half maximum (FWHM) values also increase from benzene, toluene to xylene and were found to be 39 nm, 50 nm, and 61 nm, respectively (Table 1).
For comparison, we recorded the emission spectra of the starting material 1,5-diaminonaphthalene (DAN) and the unsymmetrical 1-isocyano-5-aminonaphthalene (ICAN) in benzene, toluene and p-xylene (Fig. 1d). Surprisingly, no effect could be observed for DAN, since all three normalized spectra perfectly overlap. However, a miniscule effect is observable in the case of ICAN, and interestingly the order of bathochromic shifts is reversed, that is the emission spectrum recorded in benzene can be found at a few nm higher than those recorded in toluene or p-xylene. It may be concluded that the aromatic interaction is in connection with the electron density of the naphthalene core in the order of DIN, ICAN and DAN, showing that lower electron density is more favored for π-complex formation.
(a) The emission spectra of 1,5-diisocyanonaphthalene (1,5-DIN) in solvents of different polarity. (b) The UV-vis absorption spectra and (c) emission spectra of 1,5-DIN in benzene, toluene and p-xylene. (d) The normalized emission spectra of 1,5-diaminonaphthalene (DAN) and 1-isocyano-5-aminonaphthalene (ICAN) in benzene, toluene and p-xylene. ([DIN] = 1.25 × 10− 5 M, [DAN] = 1.197 × 10− 5 M, [ICAN] = 1.197 × 10− 5 M, T = 20 °C, λex, DIN = 317 nm, λex, DAN = 333 nm, λex, ICAN = 340 nm).
Optical characterization of 1,5-diisocyanonaphthalene (1,5-DIN) in various aromatic solvents
To gain quantitative insight into the aromatic complex formation of 1,5-DIN, UV-Vis and steady state emission spectra were recorded in a number of different aromatic solvents (Fig. 2), selected to cover a wide range of electron donating ability. It should be noted that the emission spectrum of each pure solvent was also recorded to exclude the presence of any fluorescent impurities. The emission spectra are presented in Fig. 2a–d, and the results are collected in Table 1.
It is evident from Fig. 2a that each aromatic solvent has a unique effect on the emission properties of 1,5-DIN, however, some general conclusions can also be drawn. The higher the electron density on the aromatic ring of the solvent, the higher the bathochromic shift of the emission maximum. For example, in the case of trifluorotoluene (Fig. 2a, black line), having strong EWG, no redshift and no change in the shape of the emission spectrum compared to that of in acetonitrile could be observed (Fig. 2a, dashed line). It is also apparent, analogously to Fig. 1c, that three distinct type of emission bands can be identified in Fig. 2b-d, such as benzene-, toluene- and xylene-type emissions. Benzene type emission (Fig. 1c left and Fig. 2b) can be characterized by the presence of two peaks located at 345 and 348 nm, including benzene, benzonitrile, chlorobenzene and ortho-dichlorobenzene (ODCB) as solvents. Bromo- and iodobenzene would possibly belong to this group, however, complete quenching of 1,5-DIN fluorescence could be observed most probably due to the well-known heavy atom effect31.
The second group is the toluene-type, including toluene, ethylbenzene, benzyl chloride and benzyl alcohol. The emission spectrum in this group is characterized by a broad emission band with a maximum located at 354–356 nm (Fig. 2c) and a slight shoulder at 340 nm (29 400 cm-1), which can be the remainder of the higher energy peak of the benzene type emission (Fig. 2b) at λem = 338–340 nm.
The third type emission is the xylene-type, which includes o-, m-, p-xylene and mesitylene, i.e. benzene derivatives substituted with at least two EDG as alkyl substituents. In this case the emission spectrum contains only one, strongly redshifted broad band with λem, max = 373–374 nm (26 800 − 26 740 cm-1) for mesitylene and o-xylene. Interestingly, the emission maximum of the meta isomer is found at the highest energy λem, max = 365 nm (27 400 cm-1). The highest redshift is observed for p-xylene, with λem, max = 380 nm (26 300 cm-1) as maximum, almost 10 nm higher, than that of o-xylene, indicating some kind of specific interaction between p-xylene and 1,5-DIN.

The emission spectra of 1,5-diisocyanonaphthalene (1,5-DIN) recorded in different aromatic solvents (a). The spectrum recorded in acetonitrile (MeCN, dashed line) was included for comparison. The normalized emission spectra of 1,5-diisocyanonaphthalene (1,5-DIN) recorded in aromatic solvents. ([1,5-DIN] = 1.25 × 10− 5 M, T = 20 °C, λex, DIN = 317 nm, V = 3.00 cm3).
Theoretical results
The experimental results showed a significant red shift in the emitted spectra in the presence of different xylene derivatives and mesitylene compared to other substituted benzenes, which should be studied on theoretical level, as well. This phenomenon can be explained by the formation of a strong π-π interaction between 1,5-DIN and the phenyl ring of the solvent molecule. In this way mono- (Complex-I) and bis-complexes (Complex-II) were optimized with parallel arrangement at ground (S0) and excited states (S1) states at M06-2X/6-311G++(2d,2p)[PCM(MeCN)] level of theory. The so-called T-arrangement, where the phenyl ring is perpendicular to the plane of the naphthalene, proved to be not relevant in either case. Two aspects were studied during the complexation. At first, we calculated the excitation (λex) and the emitted wavelengths (λem) of the various complexes A and B (Fig. 3; Table 2). Secondly, we calculated the enthalpies of the complex formation [ΔH(A) and ΔH(B)]. These enthalpies were corrected by an arbitrary reference complexation reaction of 1,5-DIN and hexane, which does not involve the π-π interaction [ΔΔH(A) and ΔΔH(B)]. If one studies the calculated emitted wavelengths, in all the cases the λem shifted to red by 10–20 nm for Complex-I and 20–30 nm for Complex-II. It should be mentioned, that the highest red shift is predicted for toluene, the o,m,p-xylenes and mesitylene, in agreement with the observations. However, the calculated emission for example for benzene has similar red shift such as others, contrary with the experiment. For the complete picture, we should consider the complexation enthalpies as well. The highest ΔΔH(I) and ΔΔH(II) values were exhibited by m and p-xylenes as well as Ph–Cl (Table 2). The least interaction is predicted for pyridine and benzene, which explain the observed weak red shift in these solvents.
In the structure of the π-π Complex-I, the phenyl ring placed in a non-central position above and under one of the rings of the naphthalene plane in a parallel arrangement. According to the calculated Mulliken charges separated to 1,5-DIN and the solvent molecule upon the complex formation, significant amount of negative charge is placed from the phenyl ring to the 1,5-DIN, as illustrated in Fig. 4 left side. Just as an example for Complex-I, this charge shift is − 0.032 for benzene, − 0.045 for toluene, while–0.078 for p-xylene (Fig. 4 right). This trend is in a good correlation with the general expectation that EDGs provide more electron density toward the electron deficient ring of 1,5-DIN. Upon excitation, the charge shift from phenyl ring toward naphthalene ring increases by 0.01 in all the three cases, which is considerable.
This charge transfer and the merged π-π electron system can be understood by the HOMO orbital of the Complex-I (Fig. 5). In the case of p-xylene, toluene and benzene complex, the HOMO and HOMO–1 orbitals are the strong composition of the former two HOMOs of aromatic ring as well as 1,5-DIN, referring to a merged electron system. In the first order excitation, these HOMO and HOMO-1 are involved. In contrast to the EDGs such as Me, the EWGs such as CF3 and CN result an electron deficient aromatic phenyl ring, which is already not able to provide electron density toward the also electron deficient naphthalene, which minimize the interaction between the two molecules (Fig. 5). Interestingly, in the LUMO there is no similar overlap between the two LUMOs, as shown in Fig. 5.

Top left: The π-π complex formation of 1,5-DIN with a substituted phenyl ring for Complex-I and II. Bottom left: The reference complexation reaction with non-aromatic cyclohexane. Right: Comparison of computed corrected enthalpies [ΔΔH(I); [ΔΔH(II)] for the mono and binary complexation of 1,5-DIN and some substituted phenyl derivatives at M06-2X/6-311G++(2d,2p)[PCM(MeCN)] level of theory. A = benzene; B = pyridine; C = toluene; D = o-xylene; E = m-xylene; F = p-xylene; G = mesitylene; H = Ph–CF3; I = Ph–CN; J = Ph–Cl; K = Ph–CH2OH;

Photochemical processes of Complex-I as well as II of 1,5-DIN and aromatic at M06-2X/6-311G++(2d,2p)[PCM(MeCN)] level of theory.

Left: Orbital energies and graphical representation of selected molecular orbitals (from HOMO–2 to LUMO + 2) of 1,5-DIN, benzene and its mono-complex (Complex-I) at M06-2X/6-311G++(2d,2p)[PCM(MeCN)] level of theory. Right: The HOMO and LUMO molecular orbitals.
Analytical applicability of p-complex formation
The different complexation strength and the accompanying spectral changes may be utilized in compositional analysis of the industrially very important benzene-toluene-xylene (BTX) fractions. To confirm this assumption, small amounts of p-xylene were added to the dilute solution of 1,5-DIN in benzene or toluene and fluorescence spectra were recorded using the common excitation wavelength of λex = 317 nm (Fig. 6).

Spectral changes upon the addition of p-xylene into the benzene (A) and toluene (B) solution of 1,5-DIN. The (C) figure demonstrates spectral changes when toluene is added into the benzene solution of 1,5-DIN. Each line corresponds to the addition of 10 µL of p-xylene or toluene. ([1,5-DIN] = 1.25 × 10− 5 M, T = 20 ºC, V0 = 3000 µL, λex = 317 nm).
As it is seen in Fig. 6A, the dosage of p-xylene into the benzene solution of DIN exhibits a significant concentration dependent drop of the original double peak structure at 338 and 348 nm, respectively. At the same time, the emission intensity increases at the lower energy (higher wavelength) > 380 nm region of the spectrum. The presence of one distinct isosbestic point at 369 nm indicates the presence of two chemical species, which are assumed to be the benzene and p-xylene being in complex with 1,5-DIN. Similar effects can be observed in the case of toluene-p-xylene system, where the isosbestic point is found at 375 nm. In contrast to that, when toluene was added to the benzene solution of 1,5-DIN, only the decrease of the peaks is evident. The reversed changes at lower and higher wavelengths may provide a facile ratiometric analytical method for the detection (determination) of p-xylene. The unambiguous detection of toluene may not be realized because of the lack of a selective bands. The development of a precise analytical method goes beyond the scope of this paper, it can be concluded, however that the detection limit of p-xylene in benzene is well below 0.027 M or 0.33% (v/v) as is demonstrated in Fig. 6A.








Leave a Reply